Generating Molecular Fragmentation Graphs with Autoregressive Neural Networks
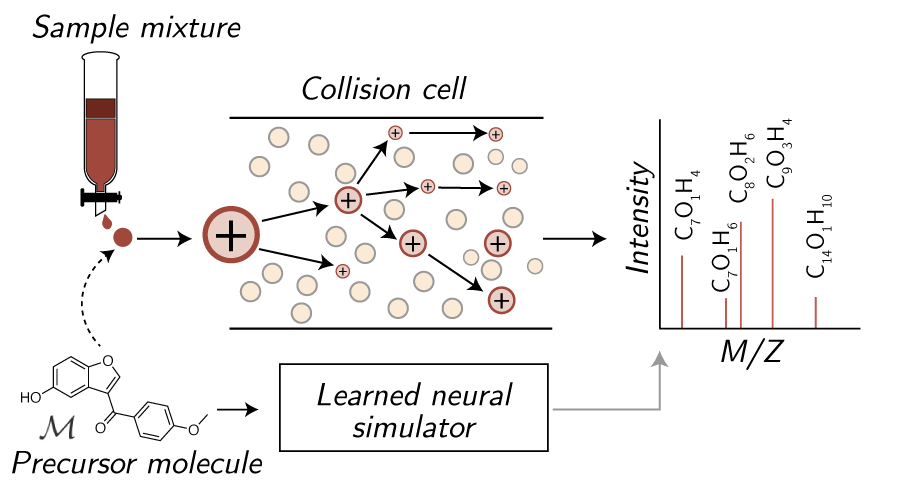
The accurate prediction of tandem mass spectra from molecular structures has the potential to unlock new metabolomic discoveries by augmenting the community's libraries of experimental reference standards. Cheminformatic spectrum prediction strategies use a "bond-breaking" framework to iteratively simulate mass spectrum fragmentations, but these methods are (a) slow, due to the need to exhaustively and combinatorially break molecules and (b) inaccurate, as they often rely upon heuristics to predict the intensity of each resulting fragment; neural network alternatives mitigate computational cost but are black-box and not inherently more accurate. We introduce a physically-grounded neural approach that learns to predict each breakage event and score the most relevant subset of molecular fragments quickly and accurately. We evaluate our model by predicting spectra from both public and private standard libraries, demonstrating that our hybrid approach offers state of the art prediction accuracy, improved metabolite identification from a database of candidates, and higher interpretability when compared to previous breakage methods and black box neural networks. The grounding of our approach in physical fragmentation events shows especially high promise for elucidating natural product molecules with more complex scaffolds.
PDF Abstract
